Hi everybody,
I still have a problem finding and selectig sequdences when searching for expressions in the description line of FASTA multisequence files in large dataset. I have for example a BLAST result with >20000 hit from bacterial proteins from the NCBI’s non-redundant protein database. However, these include something close to 2000 from the common pathogen Pseudomonas aeruginosa alone. When using the Find dialog in Jalview (include descriptions) and search for the species name, those sequences are highlighted in black but NOT selected.
What I need to do, is to sort the Pseudomonas seqs for the 3-5 different similarity goups (paralogs) and select a few representative examples each (the same for other multiply represented species). And for that I have to select and sort them to the top or bottom of the MSA.
Any ideas? Can the a “select sequence” option be incuded into the find dialog that is not a new feature?
One other point: it would be good to include an option in the Calculate → sort submenu “alphabetically” because the “by pairwise similarity” does not work for that many sequences.
Hi Arnulf - you’ve spotted a glaring hole in Jalview’s find function that we need to address for our next major release (2.12).
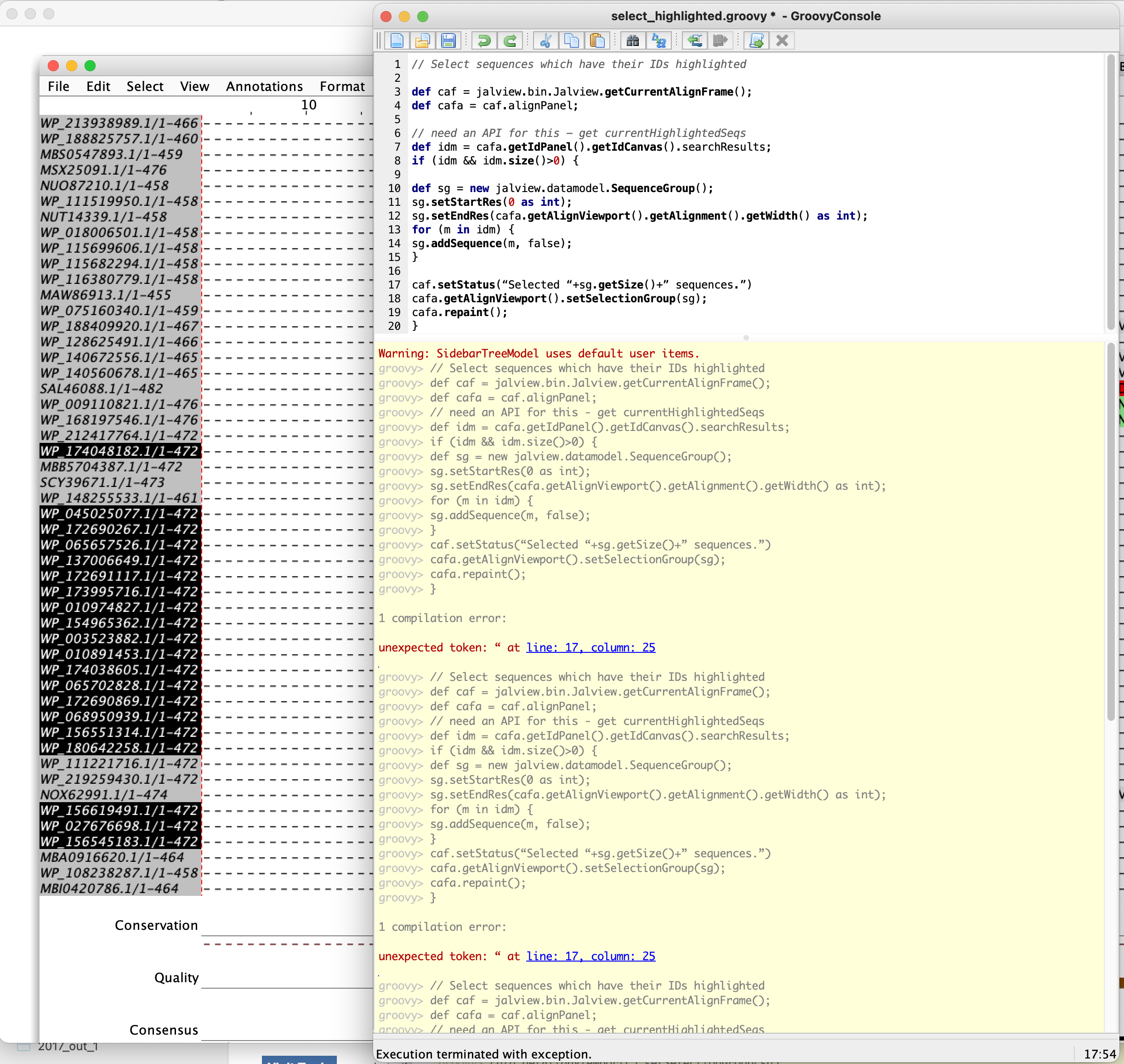
I put together a prototype for ‘select sequences from Find’ in JAL-345. Open the groovy console and paste in the following script:
// Select sequences which have their IDs highlighted
def caf = jalview.bin.Jalview.getCurrentAlignFrame();
def cafa = caf.alignPanel;
// need an API for this - get currentHighlightedSeqs
def idm = cafa.getIdPanel().getIdCanvas().searchResults;
if (idm && idm.size()>0) {
def sg = new jalview.datamodel.SequenceGroup();
sg.setStartRes(0 as int);
sg.setEndRes(cafa.getAlignViewport().getAlignment().getWidth() as int);
for (m in idm) {
sg.addSequence(m, false);
}
caf.setStatus("Selected "+sg.getSize()+" sequences.")
cafa.getAlignViewport().setSelectionGroup(sg);
cafa.repaint();
}
Just run this script to select any sequences that are highighted after - eg:
pressing the ‘Find All’ button to highlight all sequences matching the query
Adjusting the threshold in the ‘Remove Redundancy …’ dialog.
We’ll add a new function and keystroke in the Select menu to do this in 2.12 (or earlier if we manage!), but as you will see in my comments about this there are a few wrinkles that need working through to make sure the function is as useful as can be.
… happy 2022!
Jim (see additional reply to your comment about sorting below)
Actually, we’ve already got that: ‘Sort by Id’ will sort selected sequences by their identifiers.
Also: If you have structured fields in your description then you can also try the cryptically named ‘Extract Scores’ option in the calculate menu - this attempts to parse any space or tab separated values or key=value pairs in the description into sortable annotations. With any luck you’ll see a new ‘Sort by score’ submenu in the ‘Sort by’ menu that will allow you to sort according to the ‘scores’ extracted from each sequence’s description line.
many thanks for your efforts. I tried it but all seqs got selected and I got an error message (see attachment, I could not just copy the error msg from the console):
I meant sorting the sequences alphabetically, not the IDs.
I unterstand that this option sounds weird but it sorts the sequences by similarity better than the by pairwise identity option and also helps removing redundancies besides the direct command. But I should be able to do so in Excel, sorting first after species, then after sequence alphabetically and adding consecutive numbering to the sequence IDs. I would only have to think about subsequent removal of the numbering from the IDs. it is not really difficult to import medium-sized fasta sequence downloads (up to 50 000 or so) into Excel and vice versa.
The “sort by score” option sounds interesting although there are several caveats. First, the lines from Genbank and REfseq database downloads are not exactly standardized due to the number of annotation variations even of homologous proteins. Second, using the scores just from the genus/species field can help but many species have multiple non-identical and paralogous proteins from large protein families which cannot be sorted from the def lines. And third, many protein seqs from WGS and environmental databases are not annotated after species any more such as “Burkholderiales bacterium”, which encompasses many more genera besides Burkholderia.
import jalview.datamodel.*;
import jalview.analysis.*;
import jalview.commands.*;
import jalview.gui.*;
import jalview.util.QuickSort;
def af = jalview.bin.Jalview.currentAlignFrame;
def viewport = af.currentView;
def align = viewport.getAlignment();
SequenceI[] oldOrder = align.getSequencesArray();
int nSeq = align.getHeight();
String[] ids = new String[nSeq];
SequenceI[] seqs = new SequenceI[nSeq];
for (int i = 0; i < nSeq; i++)
{
// sorting by sequence string. could also add additional strings here!
ids[i] = align.getSequenceAt(i).getSequenceAsString();
seqs[i] = align.getSequenceAt(i);
}
QuickSort.sort(ids, seqs);
AlignmentSorter.setOrder(align,seqs);
af.addHistoryItem(new OrderCommand("Sort Sequences Alphabetically", oldOrder,
viewport.getAlignment()));
af.alignPanel.paintAlignment(true, false);
As the comment suggests, you could grab data from the description field if need be and prepend it to the id[i] assignment if need be.
Longer term, we’d like all these kind of functionality in Jalview 2.12. I have a prototype ‘select representatives’ routine already in development for analysing the results of HMMer searches. Your use case sounds like it has similar needs. We should have the Jalview Develop app launching Jalview 2.12-alpha in the next few weeks, so keep in contact!