I have aligned 18 sequences, and their annotated quality seems pretty good ( >70%). All but 1 sequence agrees, and that one sequence just ends early. Why is the conservation score of the pictured area so low?
annotation graph order:
Conservation score
Quality score
Consensus sequence + logo
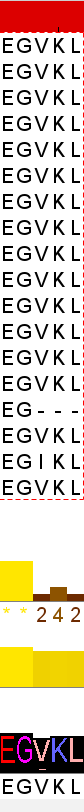
Hi !
This is an ‘historical feature’ - Jalview’s conservation score traditionally always treats gaps as another kind of conserved symbol which has all possible properties. When there more than 25% gaps, the conservation score is ‘0’ (ie it is a gap), but in this case, you see counts of just the properties shared by all amino acids at those columns (and none of the counter-properties - since gaps have all properties, but no counter-properties).
Many experts suggest Jalview should ignore gaps by default in the conservation calculation for just the reason you point out - that it seems unintuitive to report low conservation scores for columns that are otherwise identical… I added a feature to the TODO list for this (https://issues.jalview.org/browse/JAL-3046) but we haven’t implemented it yet sadly!
If this is something you’d be interested in, the patch is pretty straightforward - but this is one of those fundamental parts of Jalview where we need to do some careful testing to make sure nothing unexpected happens !
Jim.
Hi Jim,
Thanks for getting back to me. I am interested in the patch to optionally exclude gaps from conservation calculation - how would I go about implementing it?
Alex
Jalview’s source now lives over at gitlab.jalview.org. we’ve just done the migration, so the developer pages on the website still point to source.jalview.org (which just went away) - ironically you’ve given us a good reason to brush the dust off our developer onboarding documentation !
Start with a fresh clone of https://gitlab.jalview.org/jalview/jalview/ and for a new branch from ‘main’ (or releases/Release_2_11_4_Branch if you prefer).
The patch should ultimately allow configuration of the conservationThread worker in a similar way to the Consensus’ ‘ignore gaps’ option which is handled here: https://gitlab.jalview.org/jalview/jalview/-/blob/releases/Release_2_11_4_Branch/src/jalview/gui/PopupMenu.java?ref_type=heads#L1959
The actual parameter you want to change is hardcoded at https://gitlab.jalview.org/jalview/jalview/-/blob/releases/Release_2_11_4_Branch/src/jalview/workers/ConservationThread.java?ref_type=heads#L35
There are a few other wrinkles, like adding a new preference to the preferences panel, and of course tests - I’m fairly sure some existing tests will break if the parameter is changed (but haven’t actually verified that in recent memory !). If you just need something quick then those are details that can be worked on later…
I don’t think there is a straightforward answer to the gaps in a column issue. Sometimes, you might want it to show as conserved when there is a gap and other times, you want it to show as unconserved since a gap is allowed at that position.
In the original AMAS program that this feature is based on in Jalview (see this pdf)
https://www.bartongroup.org/ftp/pdf/Protein_sequence_alignments_a_strategy_for_the_hierarchical_analysis_of_residue_conservation_1993.pdf
and a server that implements the method – this was the first web server from my group – we keep it looking “retro” for this reason!
https://www.compbio.dundee.ac.uk/www-amas/
There is flexibility over how you treat gaps. AMAS is for hierarchical analysis of residue conservation and reflects the fact that sequences fall onto a tree.
Though AMAS is comprehensive, Jalview lets you explore conservation interactively. In Jalview you can subset the alignment by clustering on a tree and then look at conservation within each sub group. I find this the most useful when looking at alignments interactively since gapped sequences will typically group together and you can look at the conservation in the column in context with the hierarchical tree, then map across to structure or back to the genomic sequence.
One of the Jalview videos covers this kind of sub-family analysis on trees and I think is one of the more powerful features of Jalview for exploring column similarities and differences:
https://www.youtube.com/watch?v=8VRy9rO7Zrc
I hope all this helps?
All the best,
Geoff
–
Geoff Barton FRSE FRSB | Professor of Bioinformatics | Head of Division of Computational Biology
School of Life Sciences | University of Dundee | Scotland, UK | email: